能以千倍之速度找到潛在藥物分子的AI模型
https://bit.ly/3BlJuDM
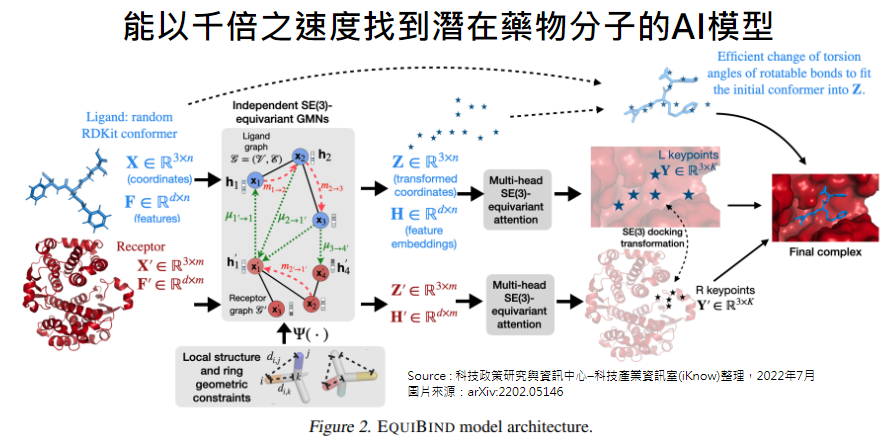
麻省理工學院的研究人員開發了一種名為EquiBind的幾何深度學習模型,比現有稱為
QuickVina2-W的計算分子嵌合模型(computational molecular docking models)快了
1,200倍的速度,成功的將類藥物的分子及蛋白質結合。
EquiBind是EquiDock的改良版,在藥物開發之前,研究人員需要找出類藥物分子並且正確
的與特定蛋白質標的綑綁及「對接」在一起。在成功與蛋白質對接後,與藥物形成配位體
(ligand),可以阻止蛋白質發揮作用。若這個現象發生在細菌的必需蛋白質(essential
protein)上,則可以殺死細菌進而保護人體。
然而藥物的發現(drug discovery)過程在財務或是計算上的成本都很高,每次藥物發現的
過程都需要投入數十億美元,並在FDA最終批准前進行了數十年的開發及測試。並且有高
達90%的藥物在進行到人體試驗階段中由於沒有任何效果或是太多副作用而失敗。製藥公
司會透過提高開發成功藥物的價格來回收這些失敗的成本。
目前找出潛在藥物候選分子計算過程的最先進計算模型依賴於大量結合評分、排名及微調
的候選本樣本,以獲得最佳蛋白質及配體的配適。透過EquiBind的模型,只需要一步就能
直接預測精準的關鍵位置,而無需事先了解蛋白質標的,即為「盲對接(blind docking)
」。
有別於大多數需要多次嘗試才能在蛋白質中的配體找到有利位置的模型,EquiBind已經內
建幾何推理(geometric reasoning)功能,可以幫助模型學習分子基本的物理特性,並成
功地廣泛對新的或未曾見過的資料進行更好的預測。
這個發現目前已經引起Relay Therapeutics的首席數據長使用該模型來進行現有用於肺癌
、白血病及腸胃道腫瘤的藥物及蛋白質的模型測試,發現EquiBind比起過去傳統的對接方
法更成功。目前研究團隊致力於了解企業專業的使用回饋,來進一步改善該模型。
該研究由Pharmaceutical Discovery and Synthesis財團、Jameel診所、DTRA
Discovery of Medical Countermeasures Against New and Emerging threats計畫、
DARPA Accelerated Molecular Discovery計畫、MIT-Takeda獎學金及NSF Expeditions
grant Collaborative Research: Understanding the World Through Code資助,並發表
在ICML研討會上。
--